ALOX15 Rabbit pAb (一抗) | Bioss
货号:bs-6505R
产品详情
相关标记
相关产品
相关文献
常见问题
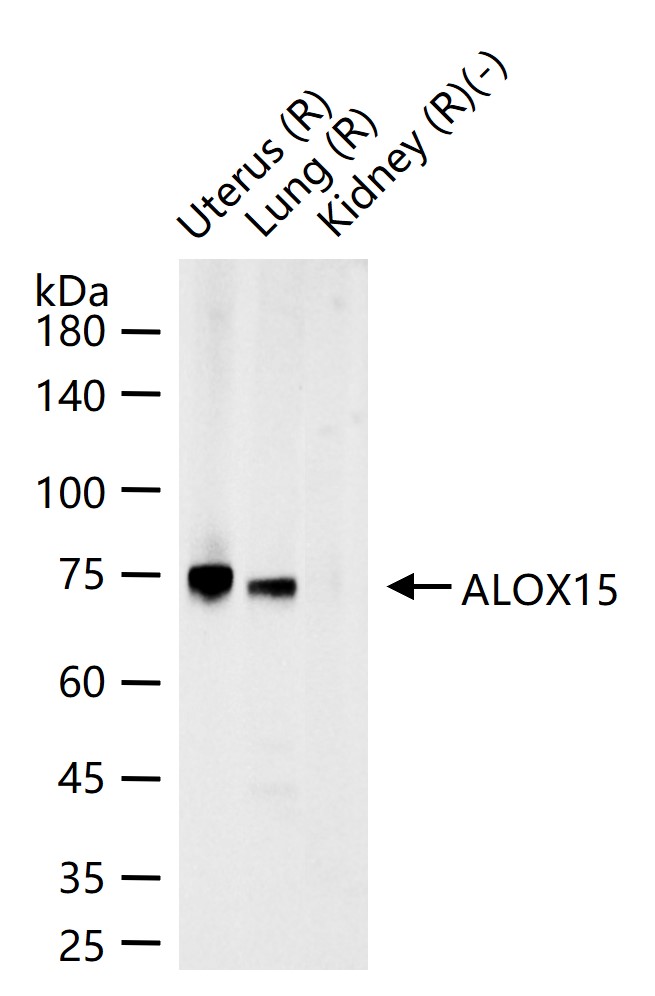
概述
产品编号
bs-6505R
英文名称
ALOX15 Rabbit pAb
中文名称
花生四烯酸15脂氧合酶1抗体
英文别名
12-LOX; 15-LOX; 15-LOX-1; LOG15; Alox12; Alox12l; LOX15_HUMAN; ALOX15; 12/15-lipoxygenase; Arachidonate 12-lipoxygenase, leukocyte-type (12-LOX); Arachidonate 15-lipoxygenase (15-LOX | 15-LOX-1); Arachidonate omega-6 lipoxygenase; Hepoxilin A3 synthase Alox15; Linoleate 13S-lipoxygenase; LOX15_RAT; Arachidonate 15-lipoxygenase (15-LOX); arachidonate 15-lipoxygenase
抗体来源
Rabbit
免疫原
KLH conjugated synthetic peptide derived from mouse ALOX15/15 Lipoxygenase 1: 581-662/662
亚型
IgG
性状
Liquid
纯化方法
affinity purified by Protein A
克隆类型
Polyclonal
理论分子量
73 kDa
检测分子量
75 kDa
浓度
1mg/ml
储存液
0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol.
研究领域
Cardiovascular > Atherosclerosis > Lipoprotein metabolism
Cardiovascular > Lipids / Lipoproteins > Fatty Acids > Metabolism
Metabolism > Pathways and Processes > Metabolic signaling pathways > Lipid and lipoprotein metabolism > Fatty acids
Metabolism > Pathways and Processes > Metabolic signaling pathways > Lipid and lipoprotein metabolism > Lipoprotein metabolism
Metabolism > Pathways and Processes > Redox metabolism > Fatty acid oxidation
保存条件
Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles.
注意事项
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
数据库链接
背景资料
Lipoxygenases are a family of enzymes which dioxygenate unsaturated fatty acids, thus initiating lipoperoxidation of membranes and synthesis of signaling molecules, as well as inducing structural and metabolic changes in the cell. The Lox enzymes in mammals include 12-LO and 15-LO, which are classified with respect to their positional specificity of the deoxygenation of their most common substrate, arachidonic acid. The metabolism of arachidonic acid leads to the generation of biologically active metabolites that have been implicated in cell growth and proliferation, as well as survival and apoptosis. 15-Lipoxygenase (15-LO) acts in physiological membrane remodeling and the pathogenesis of atherosclerosis, inflammation, and carcinogenesis. It is highly regulated and expressed in a tissue- and cell-type-specific fashion. IL-4 and IL-13 play important roles in transactivating the 15-LO gene. Overexpression of 15-LO type 1 in prostate cancer contributes to the cancer progression by regulating IGF-1R expression and activation.
产品应用
| 应用 | 已检合格种属 | 预测种属 | 推荐稀释比例 |
|---|---|---|---|
| WB | Rat | 1:500-2000 |
交叉反应
交叉反应: Rat
相关产品
暂无相关产品
靶标
基因名
ALOX15
蛋白名
Polyunsaturated fatty acid lipoxygenase ALOX15
亚基
Homotetramer. Can also form heterotetramers with RYR2. Interacts with CALM; CALM with bound calcium inhibits the RYR1 channel activity. Interacts with S100A1. Interacts with FKBP1A; this stabilizes the closed conformation of the channel. Interacts with CACNA1S; interaction with CACNA1S is important for activation of the RYR1 channel. Interacts with CACNB1. Interacts with TRDN and ASPH; these interactions stimulate RYR1 channel activity (By similarity). Identified in a complex composed of RYR1, PDE4D, PKA, FKBP1A and protein phosphatase 1 (PP1). Repeated very high-level exercise decreases interaction with PDE4D and protein phosphatase 1 (PP1).
亚细胞定位
Cytoplasm.
组织特异性
Skeletal muscle and brain (cerebellum and hippocampus).
翻译后修饰
Channel activity is modulated by phosphorylation. Phosphorylation at Ser-2843 may increase channel activity. Repeated very high-level exercise increases phosphorylation at Ser-2843.
Activated by reversible S-nitrosylation. Repeated very high-level exercise increases S-nitrosylation.
Activated by reversible S-nitrosylation. Repeated very high-level exercise increases S-nitrosylation.
疾病
Malignant hyperthermia 1 (MHS1) [MIM:145600]: Autosomal dominant pharmacogenetic disorder of skeletal muscle and is one of the main causes of death due to anesthesia. In susceptible people, an MH episode can be triggered by all commonly used inhalational anesthetics such as halothane and by depolarizing muscle relaxants such as succinylcholine. The clinical features of the myopathy are hyperthermia, accelerated muscle metabolism, contractures, metabolic acidosis, tachycardia and death, if not treated with the postsynaptic muscle relaxant, dantrolene. Susceptibility to MH can be determined with the 'in vitro' contracture test (IVCT): observing the magnitude of contractures induced in strips of muscle tissue by caffeine alone and halothane alone. Patients with normal response are MH normal (MHN), those with abnormal response to caffeine alone or halothane alone are MH equivocal (MHE(C) and MHE(H) respectively). Note=The disease is caused by mutations affecting the gene represented in this entry.
Central core disease of muscle (CCD) [MIM:117000]: Autosomal dominant congenital myopathy, but a severe autosomal recessive form also exists. Both clinical and histological variability is observed. Affected individuals typically display hypotonia and proximal muscle weakness in infancy, leading to the delay of motor milestones. The clinical course of the disorder is usually slow or nonprogressive in adulthood, and the severity of the symptoms may vary from normal to significant muscle weakness. Microscopic examination of CCD-affected skeletal muscle reveals a predominance of type I fibers containing amorphous-looking areas (cores) that do not stain with oxidative and phosphorylase histochemical techniques. Note=The disease is caused by mutations affecting the gene represented in this entry.
Multiminicore disease with external ophthalmoplegia (MMDO) [MIM:255320]: Clinically heterogeneous neuromuscular disorder. General features include neonatal hypotonia, delayed motor development, and generalized muscle weakness and amyotrophy, which may progress slowly or remain stable. Muscle biopsy shows multiple, poorly circumscribed, short areas of sarcomere disorganization and mitochondria depletion (areas termed minicores) in most muscle fibers. Typically, no dystrophic signs, such as muscle fiber necrosis or regeneration or significant endomysial fibrosis, are present in multiminicore disease. Note=The disease is caused by mutations affecting the gene represented in this entry.
Congenital myopathy with fiber-type disproportion (CFTD) [MIM:255310]: Genetically heterogeneous disorder in which there is relative hypotrophy of type 1 muscle fibers compared to type 2 fibers on skeletal muscle biopsy. However, these findings are not specific and can be found in many different myopathic and neuropathic conditions. Note=The disease is caused by mutations affecting the gene represented in this entry.
Note=Defects in RYR1 may be a cause of Samaritan myopathy, a congenital myopathy with benign course. Patients display severe hypotonia and respiratory distress at birth. Unlike other congenital myopathies, the health status constantly improves and patients are minimally affected at adulthood.
Central core disease of muscle (CCD) [MIM:117000]: Autosomal dominant congenital myopathy, but a severe autosomal recessive form also exists. Both clinical and histological variability is observed. Affected individuals typically display hypotonia and proximal muscle weakness in infancy, leading to the delay of motor milestones. The clinical course of the disorder is usually slow or nonprogressive in adulthood, and the severity of the symptoms may vary from normal to significant muscle weakness. Microscopic examination of CCD-affected skeletal muscle reveals a predominance of type I fibers containing amorphous-looking areas (cores) that do not stain with oxidative and phosphorylase histochemical techniques. Note=The disease is caused by mutations affecting the gene represented in this entry.
Multiminicore disease with external ophthalmoplegia (MMDO) [MIM:255320]: Clinically heterogeneous neuromuscular disorder. General features include neonatal hypotonia, delayed motor development, and generalized muscle weakness and amyotrophy, which may progress slowly or remain stable. Muscle biopsy shows multiple, poorly circumscribed, short areas of sarcomere disorganization and mitochondria depletion (areas termed minicores) in most muscle fibers. Typically, no dystrophic signs, such as muscle fiber necrosis or regeneration or significant endomysial fibrosis, are present in multiminicore disease. Note=The disease is caused by mutations affecting the gene represented in this entry.
Congenital myopathy with fiber-type disproportion (CFTD) [MIM:255310]: Genetically heterogeneous disorder in which there is relative hypotrophy of type 1 muscle fibers compared to type 2 fibers on skeletal muscle biopsy. However, these findings are not specific and can be found in many different myopathic and neuropathic conditions. Note=The disease is caused by mutations affecting the gene represented in this entry.
Note=Defects in RYR1 may be a cause of Samaritan myopathy, a congenital myopathy with benign course. Patients display severe hypotonia and respiratory distress at birth. Unlike other congenital myopathies, the health status constantly improves and patients are minimally affected at adulthood.
相似性
Belongs to the lipoxygenase family.
Contains 1 lipoxygenase domain.
Contains 1 PLAT domain.
Contains 1 lipoxygenase domain.
Contains 1 PLAT domain.
功能
Oxygenase and 14,15-leukotriene A4 synthase activity. Converts arachidonic acid to 15S-hydroperoxyeicosatetraenoic acid. Also acts on C-12 of arachidonate as well as on linoleic acid.
同靶标产品
相关文献
提示: 发表研究结果有使用 bs-6505R 时请让我们知道,以便我们可以引用参考文章。作为回馈,资料提供者将获得我们送上的小礼品。
具体参考文献:bs-6505R 被引用于8文献中