Collagen I Recombinant Rabbit mAb (一抗) - IHC-P,IHC-F,IF,Flow-Cyt | Bioss
Rrmab?兔单抗
货号:bsm-52478R
产品详情
相关标记
相关产品
相关文献
常见问题
概述
产品编号
bsm-52478R
产品类型
重组兔单抗、农牧业/家禽抗体、mIHC精品抗体
英文名称
Collagen I Recombinant Rabbit mAb
中文名称
I型胶原重组兔单抗
英文别名
CAFYD; EDSARTH1; EDSC; OI1; OI2; OI3; OI4; Col1a-1; Cola-1; Cola1; Mov-13; Mov13; CO1A1_BOVIN; COL1A1; Alpha-1 type I collagen; CO1A2_CANLF; COL1A2; Alpha-2 type I collagen; collagen type I alpha 1 chain; collagen, type I, alpha 1
抗体来源
Rabbit
免疫原
KLH conjugated synthetic peptide derived from human Collagen I
亚型
IgG
性状
Liquid
纯化方法
affinity purified by Protein A
克隆类型
Recombinant
克隆号
5F2
理论分子量
130 kDa
浓度
1mg/ml
储存液
0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol.
研究领域
SWISS
Gene ID
保存条件
Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles.
注意事项
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
数据库链接
产品介绍
I型胶原是软骨基质中的一种结构蛋白 。
主要用于Ⅰ型胶原分布及变态反应方面的研究。
背景资料
Collagens are highly conserved throughout evolution and are characterised by an uninterrupted "Glycine X Y" triplet repeat that is a necessary part of the triple helical structure. Type I collagen (95 kDa) is found in bone, cornea, skin and tendon. Mutations in the encoding gene are associated with osteogenesis imperfecta, Ehlers Danlos syndrome, and idiopathic osteoporosis. Reciprocal translocations between chromosomes 17 and 22, where this gene and the gene for Platelet-derived growth factor beta are located, are associated with a particular type of skin tumor called dermatofibrosarcoma protuberans, resulting from unregulated expression of the growth factor.
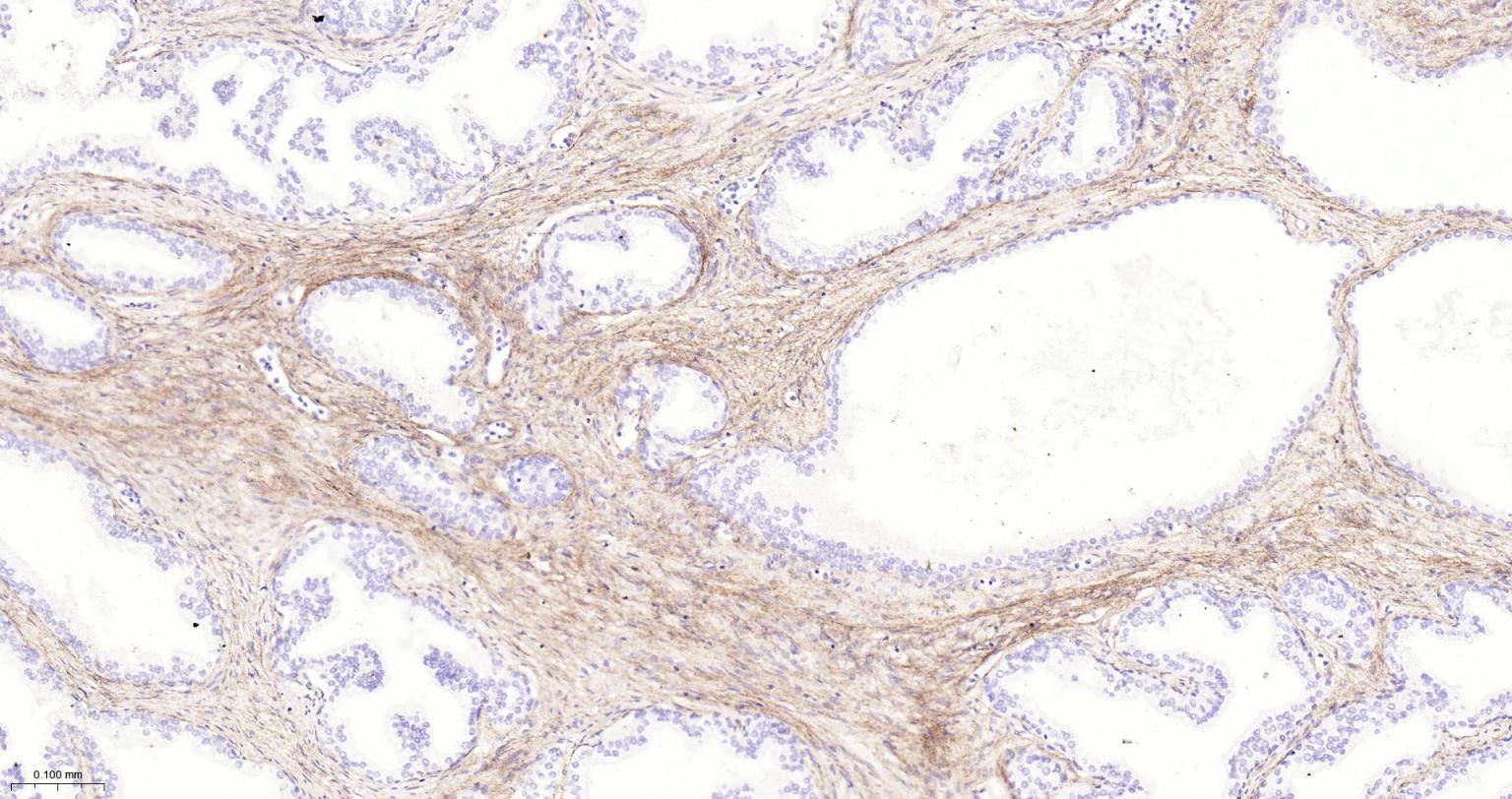
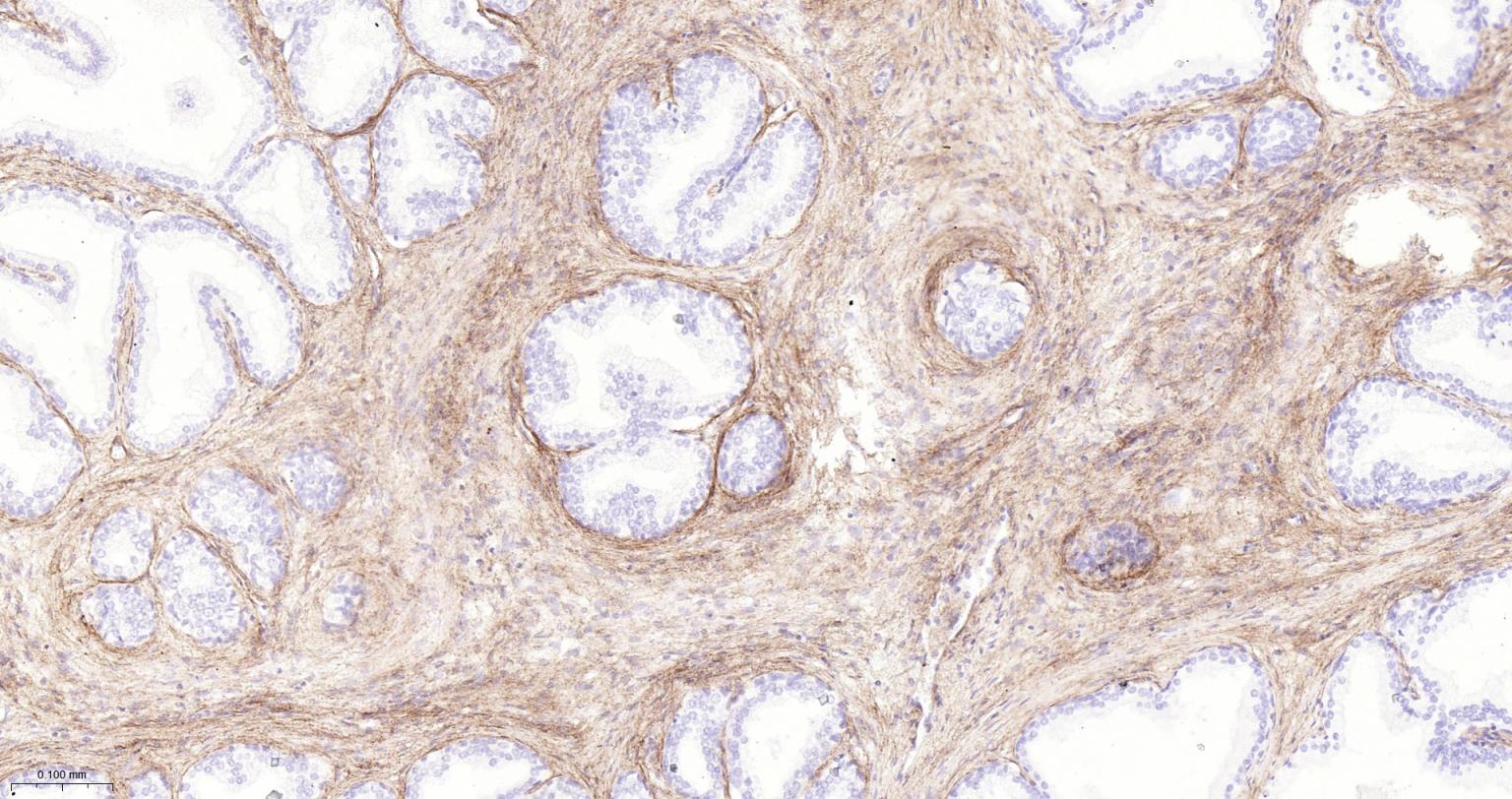
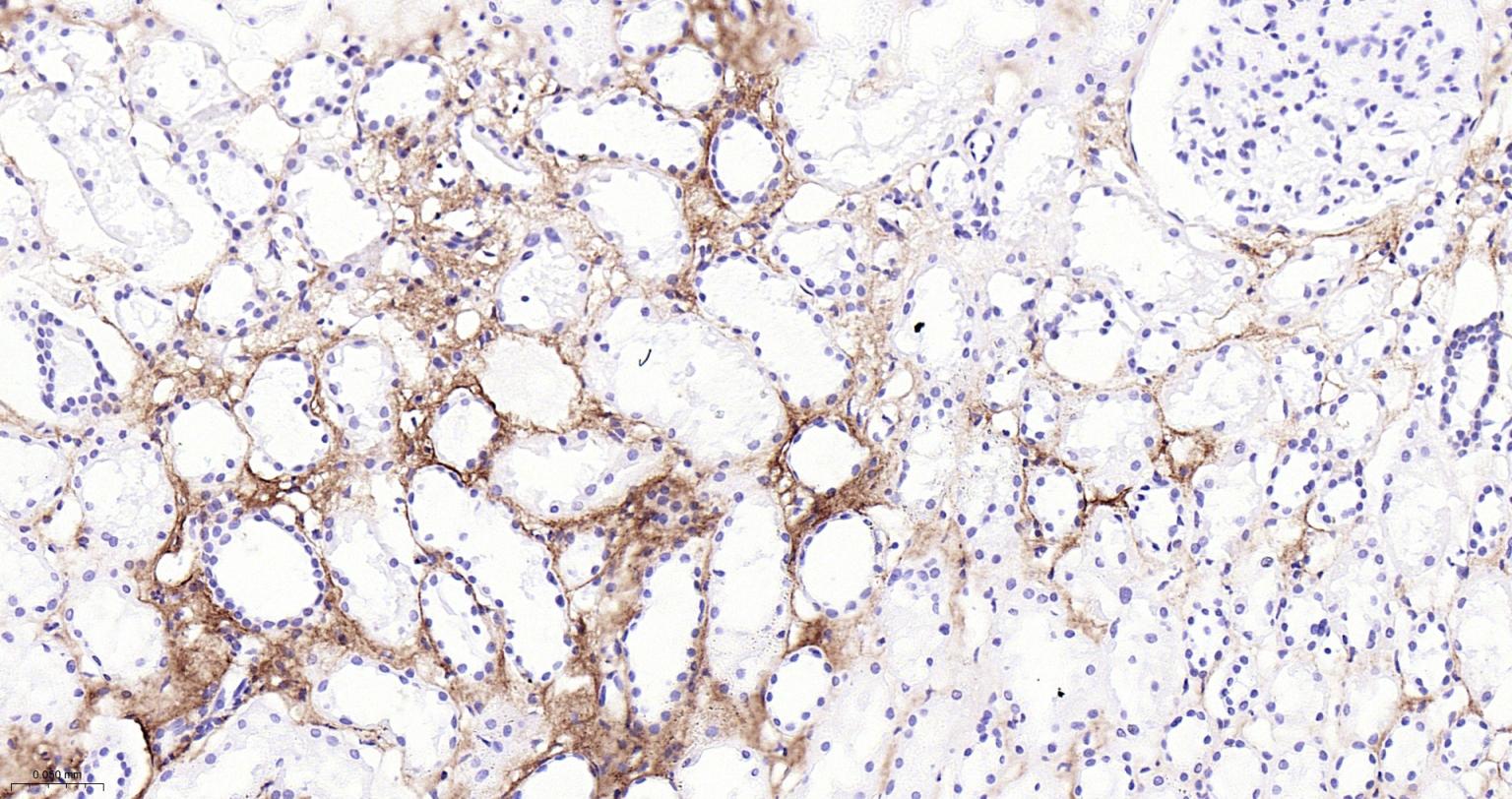
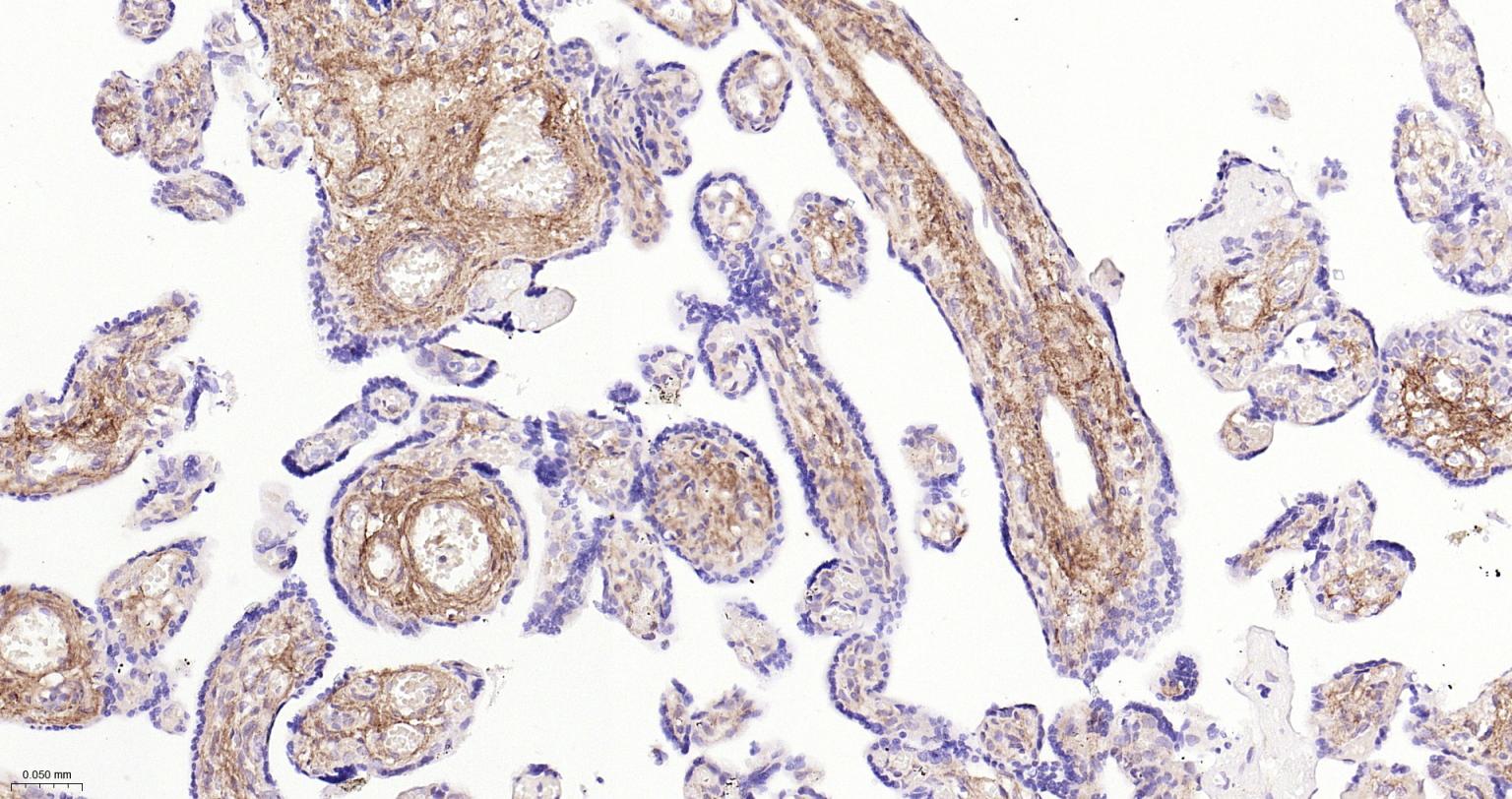
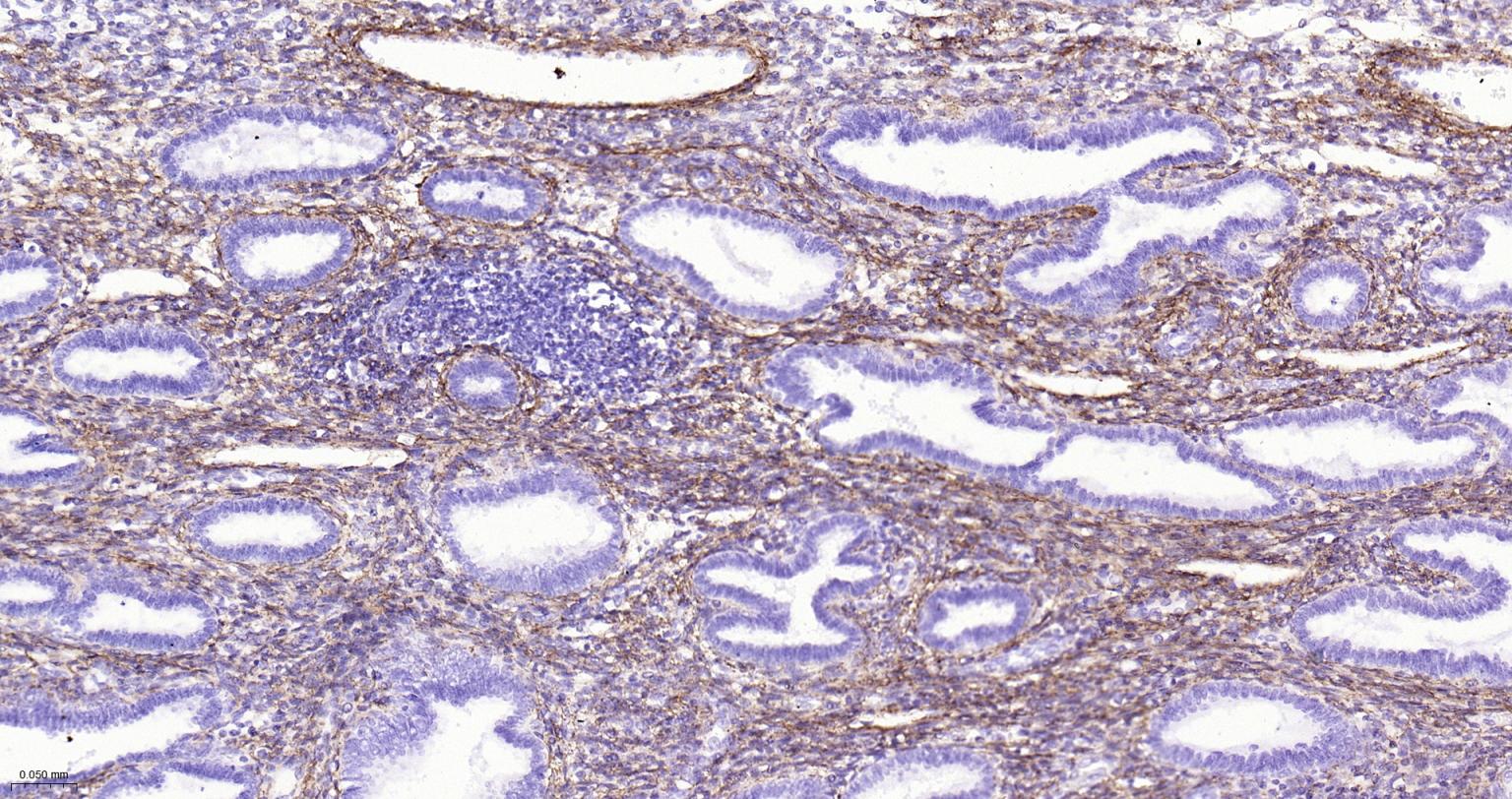
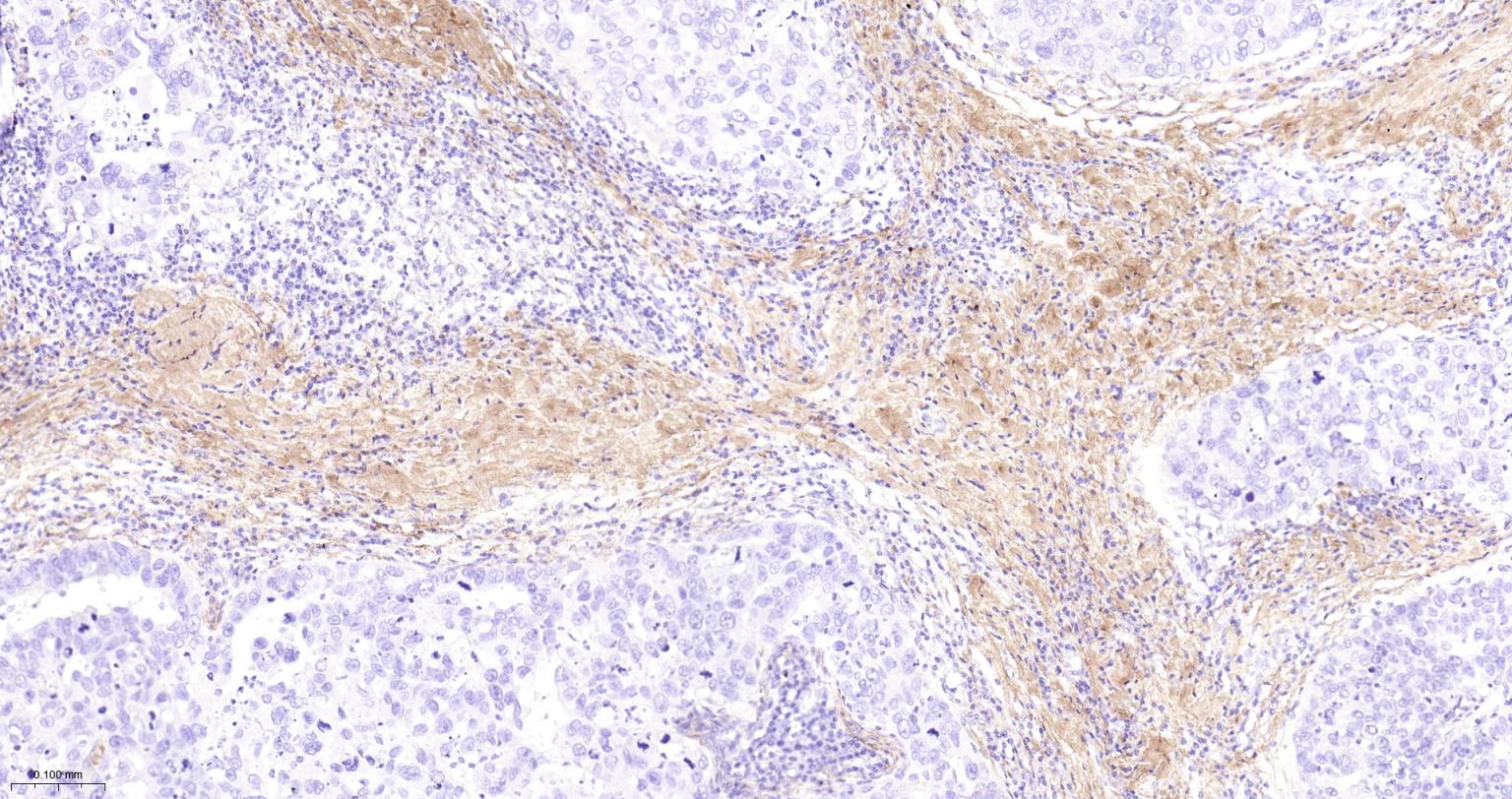
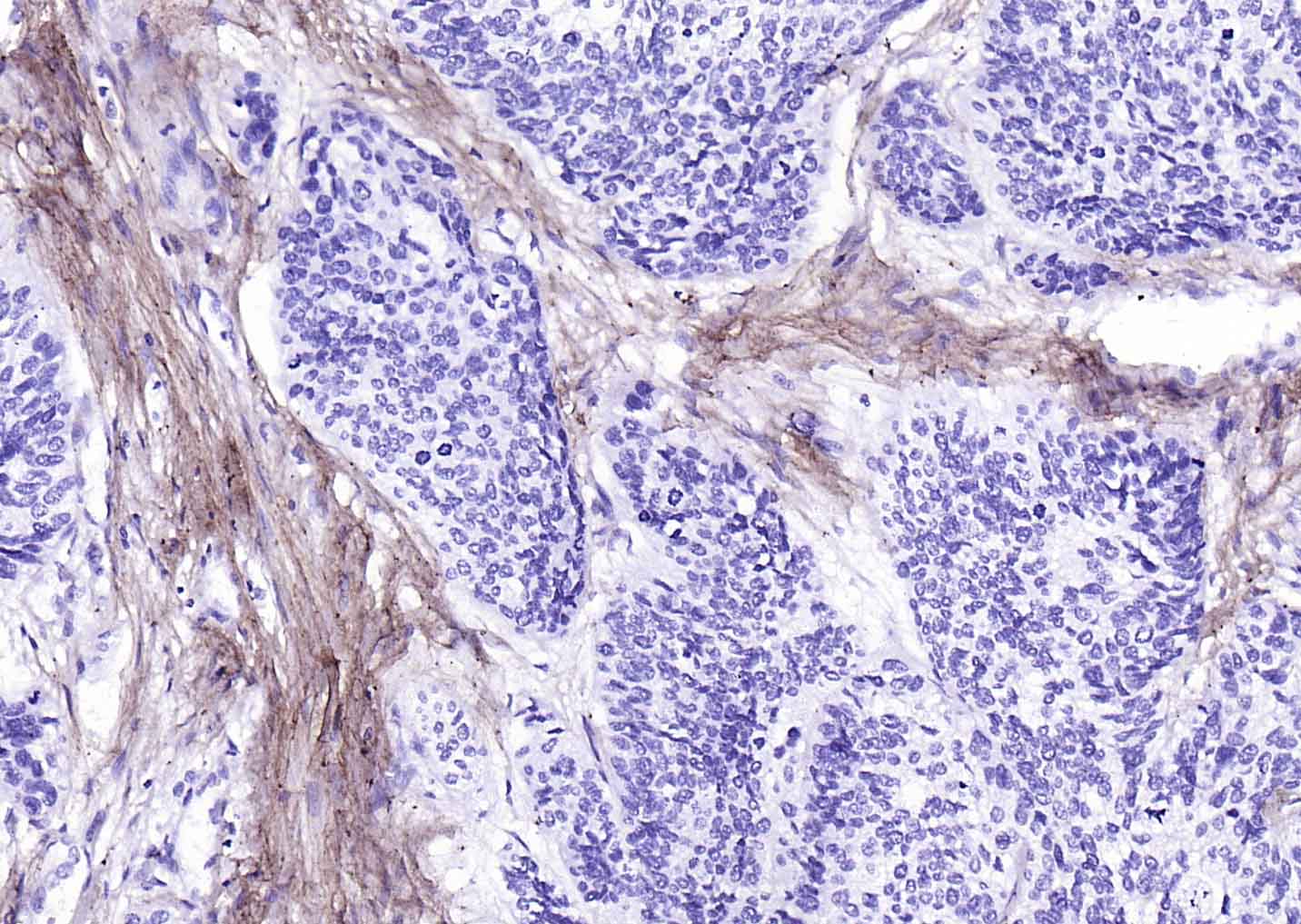
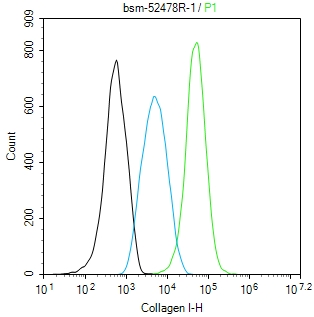
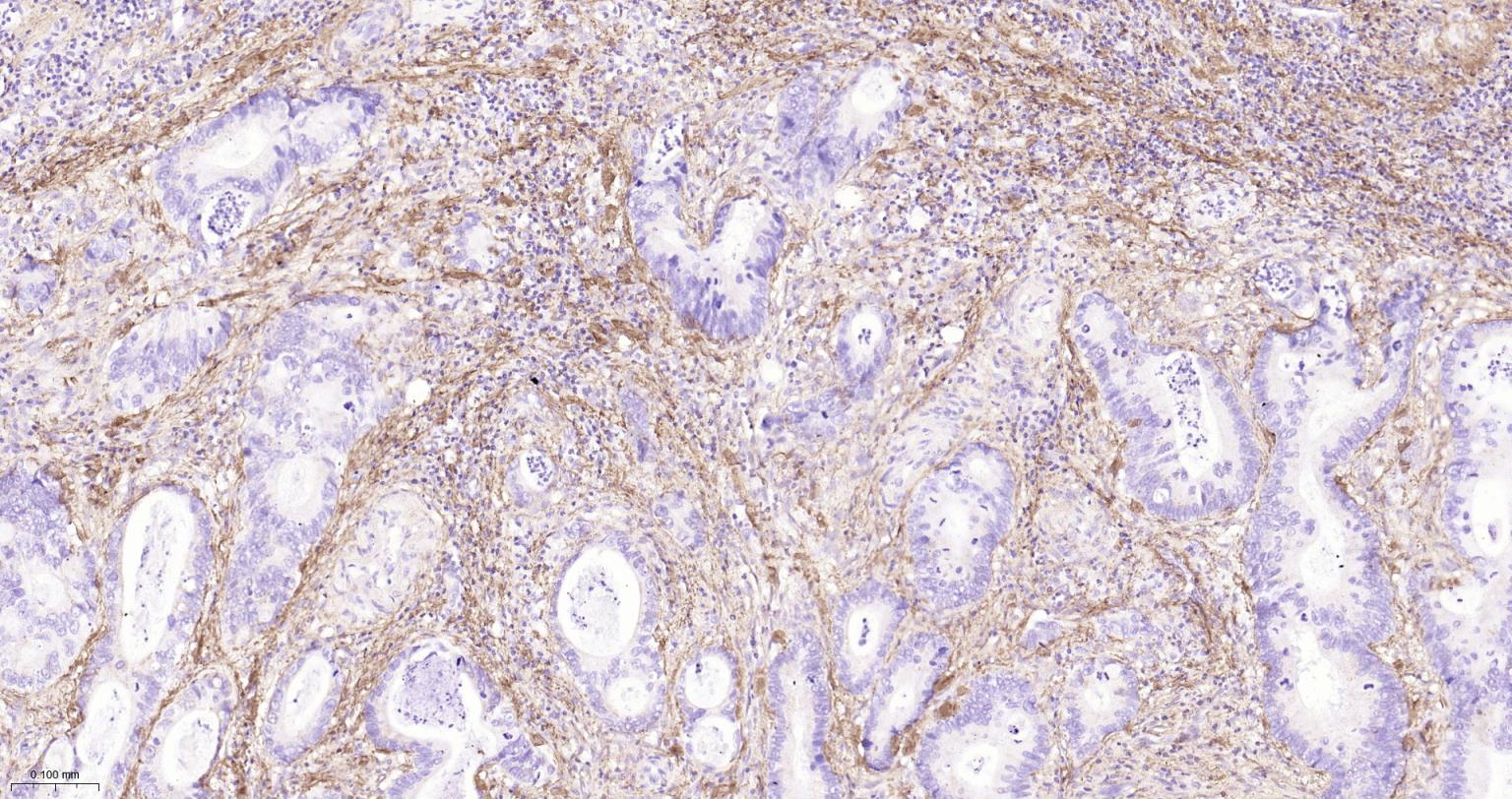
产品应用
| 应用 | 已检合格种属 | 预测种属 | 推荐稀释比例 |
|---|---|---|---|
| IHC-P | Human | 1:100-500 | |
| IHC-F | Human | 1:100-500 | |
| IF | Human | 1:100-500 | |
| Flow-Cyt | Human | 1:50-100 |
交叉反应
交叉反应: Human
相关产品
暂无相关产品
靶标
基因名
COL1A1
蛋白名
Collagen alpha-1(I) chain
亚基
Trimers of one alpha 2(I) and two alpha 1(I) chains. Interacts with MRC2. Interacts with TRAM2.
Subcellular Location : Secreted, extracellular space, extracellular matrix.
亚细胞定位
Secreted, extracellular space, extracellular matrix.
组织特异性
Forms the fibrils of tendon, ligaments and bones. In bones the fibrils are mineralized with calcium hydroxyapatite.
翻译后修饰
Proline residues at the third position of the tripeptide repeating unit (G-X-P) are hydroxylated in some or all of the chains. Proline residues at the second position of the tripeptide repeating unit (G-P-X) are hydroxylated in some of the chains.
O-linked glycan consists of a Glc-Gal disaccharide bound to the oxygen atom of a post-translationally added hydroxyl group.
O-linked glycan consists of a Glc-Gal disaccharide bound to the oxygen atom of a post-translationally added hydroxyl group.
疾病
Defects in COL1A1 are the cause of Caffey disease (CAFFD) [MIM:114000]; also known as infantile cortical hyperostosis. Caffey disease is characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age.
Defects in COL1A1 are a cause of Ehlers-Danlos syndrome type 1 (EDS1) [MIM:130000]; also known as Ehlers-Danlos syndrome gravis. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS1 is the severe form of classic Ehlers-Danlos syndrome.
Defects in COL1A1 are the cause of Ehlers-Danlos syndrome type 7A (EDS7A) [MIM:130060]; also known as autosomal dominant Ehlers-Danlos syndrome type VII. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS7A is marked by bilateral congenital hip dislocation, hyperlaxity of the joints, and recurrent partial dislocations.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 1 (OI1) [MIM:166200]. A dominantly inherited connective tissue disorder characterized by bone fragility and blue sclerae. Osteogenesis imperfecta type 1 is non-deforming with normal height or mild short stature, and no dentinogenesis imperfecta.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 2 (OI2) [MIM:166210]; also known as osteogenesis imperfecta congenita. A connective tissue disorder characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 3 (OI3) [MIM:259420]. A connective tissue disorder characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera, and dentinogenesis imperfecta.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 4 (OI4) [MIM:166220]; also known as osteogenesis imperfecta with normal sclerae. A connective tissue disorder characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta.
Genetic variations in COL1A1 are a cause of susceptibility to osteoporosis (OSTEOP) [MIM:166710]; also known as involutional or senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mass, disruption of bone microarchitecture without alteration in the composition of bone. Osteoporotic bones are more at risk of fracture.
Note=A chromosomal aberration involving COL1A1 is found in dermatofibrosarcoma protuberans. Translocation t(17;22)(q22;q13) with PDGF.
Defects in COL1A1 are a cause of Ehlers-Danlos syndrome type 1 (EDS1) [MIM:130000]; also known as Ehlers-Danlos syndrome gravis. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS1 is the severe form of classic Ehlers-Danlos syndrome.
Defects in COL1A1 are the cause of Ehlers-Danlos syndrome type 7A (EDS7A) [MIM:130060]; also known as autosomal dominant Ehlers-Danlos syndrome type VII. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS7A is marked by bilateral congenital hip dislocation, hyperlaxity of the joints, and recurrent partial dislocations.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 1 (OI1) [MIM:166200]. A dominantly inherited connective tissue disorder characterized by bone fragility and blue sclerae. Osteogenesis imperfecta type 1 is non-deforming with normal height or mild short stature, and no dentinogenesis imperfecta.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 2 (OI2) [MIM:166210]; also known as osteogenesis imperfecta congenita. A connective tissue disorder characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 3 (OI3) [MIM:259420]. A connective tissue disorder characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera, and dentinogenesis imperfecta.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 4 (OI4) [MIM:166220]; also known as osteogenesis imperfecta with normal sclerae. A connective tissue disorder characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta.
Genetic variations in COL1A1 are a cause of susceptibility to osteoporosis (OSTEOP) [MIM:166710]; also known as involutional or senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mass, disruption of bone microarchitecture without alteration in the composition of bone. Osteoporotic bones are more at risk of fracture.
Note=A chromosomal aberration involving COL1A1 is found in dermatofibrosarcoma protuberans. Translocation t(17;22)(q22;q13) with PDGF.
相似性
Belongs to the fibrillar collagen family.
Contains 1 fibrillar collagen NC1 domain.
Contains 1 VWFC domain.
Contains 1 fibrillar collagen NC1 domain.
Contains 1 VWFC domain.
功能
Type I collagen is a member of group I collagen (fibrillar forming collagen).
同靶标产品
相关文献
提示: 发表研究结果有使用 bsm-52478R 时请让我们知道,以便我们可以引用参考文章。作为回馈,资料提供者将获得我们送上的小礼品。
具体参考文献:bsm-52478R 被引用于6文献中