APOA1 Recombinant Rabbit mAb (一抗) - WB,IHC-P,IHC-F,IF | Bioss
Rrmab?兔单抗
货号:bsm-61232R
产品详情
相关标记
相关产品
相关文献
常见问题
概述
产品编号
bsm-61232R
产品类型
重组兔单抗
英文名称
APOA1 Recombinant Rabbit mAb
中文名称
载脂蛋白A1重组兔单抗
英文别名
AMYLD3; HPALP2; apo(a); APOA1_HUMAN; APOA1; Apo-AI; ApoA-I; Apolipoprotein A1; apolipoprotein A-I
抗体来源
Rabbit
免疫原
A synthesized peptide derived from human Apolipoprotein A1: 13-50
亚型
IgG
性状
Liquid
纯化方法
affinity purified by Protein A
克隆类型
Recombinant
克隆号
9B13
理论分子量
31 kDa
检测分子量
25 kDa
储存液
10mM phosphate buffered saline(pH 7.4) with 150mM sodium chloride, 0.05% BSA, 0.02% Proclin300 and 50% glycerol.
SWISS
Gene ID
保存条件
Store at 4℃ for short term. Store at -20℃ for long term. Avoid repeated freeze/thaw cycles.
注意事项
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
产品介绍
APOA1是检测高密度脂蛋白的主要形式,APOA1降低被认为是心、脑血管疾病的危险因素。
APOA1升高:常见于肝脏疾病、肝外胆道阻塞、人工透析。
APOA1降低:常见于冠心病、未控制的糖尿病、肾病综合征、营养不良、活动性肝炎、肝功能低下、动脉粥样硬化、高脂蛋白血症。
APOA1升高:常见于肝脏疾病、肝外胆道阻塞、人工透析。
APOA1降低:常见于冠心病、未控制的糖尿病、肾病综合征、营养不良、活动性肝炎、肝功能低下、动脉粥样硬化、高脂蛋白血症。
背景资料
Participates in the reverse transport of cholesterol from tissues to the liver for excretion by promoting cholesterol efflux from tissues and by acting as a cofactor for the lecithin cholesterol acyltransferase (LCAT). As part of the SPAP complex, activates spermatozoa motility.
基因名
APOA1
蛋白名
Apolipoprotein A-I
亚基
Interacts with APOA1BP and CLU. Component of a sperm activating protein complex (SPAP), consisting of APOA1, an immunoglobulin heavy chain, an immunoglobulin light chain and albumin. Interacts with NDRG1.
亚细胞定位
Secreted.
组织特异性
Major protein of plasma HDL, also found in chylomicrons. Synthesized in the liver and small intestine.
翻译后修饰
Palmitoylated.
Phosphorylation sites are present in the extracelllular medium.
Phosphorylation sites are present in the extracelllular medium.
疾病
Defects in APOA1 are a cause of high density lipoprotein deficiency type 2 (HDLD2) [MIM:604091]; also known as familial hypoalphalipoproteinemia (FHA). Inheritance is autosomal dominant.
Defects in APOA1 are a cause of the low HDL levels observed in high density lipoprotein deficiency type 1 (HDLD1) [MIM:205400]; also known as analphalipoproteinemia or Tangier disease (TGD). HDLD1 is a recessive disorder characterized by the absence of plasma HDL, accumulation of cholesteryl esters, premature coronary artery disease, hepatosplenomegaly, recurrent peripheral neuropathy and progressive muscle wasting and weakness. In HDLD1 patients, ApoA-I fails to associate with HDL probably because of the faulty conversion of pro-ApoA-I molecules into mature chains, either due to a defect in the converting enzyme activity or a specific structural defect in Tangier ApoA-I.
Defects in APOA1 are the cause of amyloid polyneuropathy-nephropathy Iowa type (AMYLIOWA) [MIM:107680]; also known as amyloidosis van Allen type or familial amyloid polyneuropathy type III. AMYLIOWA is a hereditary generalized amyloidosis due to deposition of amyloid mainly constituted by apolipoprotein A1. The clinical picture is dominated by neuropathy in the early stages of the disease and nephropathy late in the course. Death is due in most cases to renal amyloidosis. Severe peptic ulcer disease can occurr in some and hearing loss is frequent. Cataracts is present in several, but vitreous opacities are not observed. Defects in APOA1 are a cause of amyloidosis type 8 (AMYL8) [MIM:105200]; also known as systemic non-neuropathic amyloidosis or Ostertag-type amyloidosis. AMYL8 is a hereditary generalized amyloidosis due to deposition of apolipoprotein A1, fibrinogen and lysozyme amyloids. Viscera are particularly affected. There is no involvement of the nervous system. Clinical features include renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash.
Defects in APOA1 are a cause of the low HDL levels observed in high density lipoprotein deficiency type 1 (HDLD1) [MIM:205400]; also known as analphalipoproteinemia or Tangier disease (TGD). HDLD1 is a recessive disorder characterized by the absence of plasma HDL, accumulation of cholesteryl esters, premature coronary artery disease, hepatosplenomegaly, recurrent peripheral neuropathy and progressive muscle wasting and weakness. In HDLD1 patients, ApoA-I fails to associate with HDL probably because of the faulty conversion of pro-ApoA-I molecules into mature chains, either due to a defect in the converting enzyme activity or a specific structural defect in Tangier ApoA-I.
Defects in APOA1 are the cause of amyloid polyneuropathy-nephropathy Iowa type (AMYLIOWA) [MIM:107680]; also known as amyloidosis van Allen type or familial amyloid polyneuropathy type III. AMYLIOWA is a hereditary generalized amyloidosis due to deposition of amyloid mainly constituted by apolipoprotein A1. The clinical picture is dominated by neuropathy in the early stages of the disease and nephropathy late in the course. Death is due in most cases to renal amyloidosis. Severe peptic ulcer disease can occurr in some and hearing loss is frequent. Cataracts is present in several, but vitreous opacities are not observed. Defects in APOA1 are a cause of amyloidosis type 8 (AMYL8) [MIM:105200]; also known as systemic non-neuropathic amyloidosis or Ostertag-type amyloidosis. AMYL8 is a hereditary generalized amyloidosis due to deposition of apolipoprotein A1, fibrinogen and lysozyme amyloids. Viscera are particularly affected. There is no involvement of the nervous system. Clinical features include renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash.
相似性
Belongs to the apolipoprotein A1/A4/E family.
功能
Participates in the reverse transport of cholesterol from tissues to the liver for excretion by promoting cholesterol efflux from tissues and by acting as a cofactor for the lecithin cholesterol acyltransferase (LCAT). As part of the SPAP complex, activates spermatozoa motility.
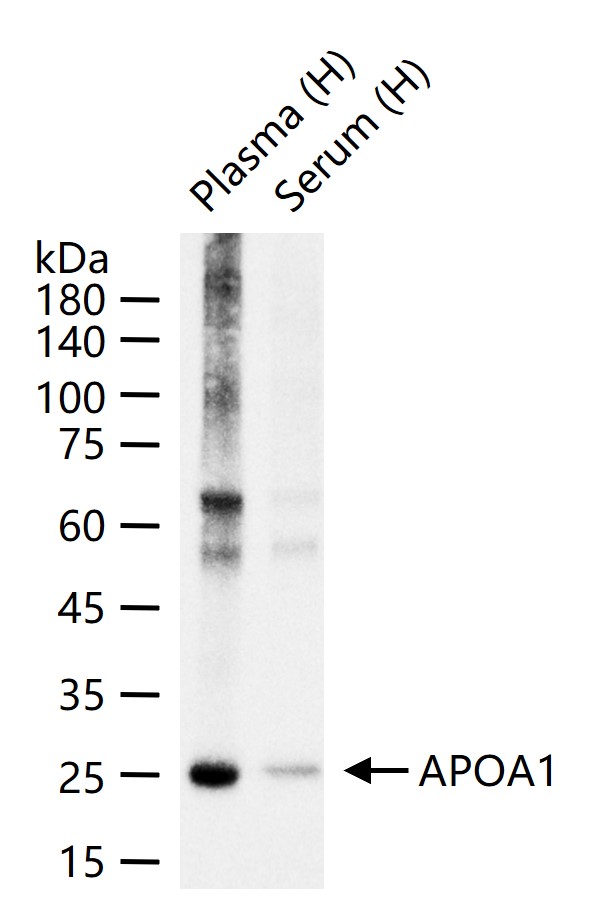
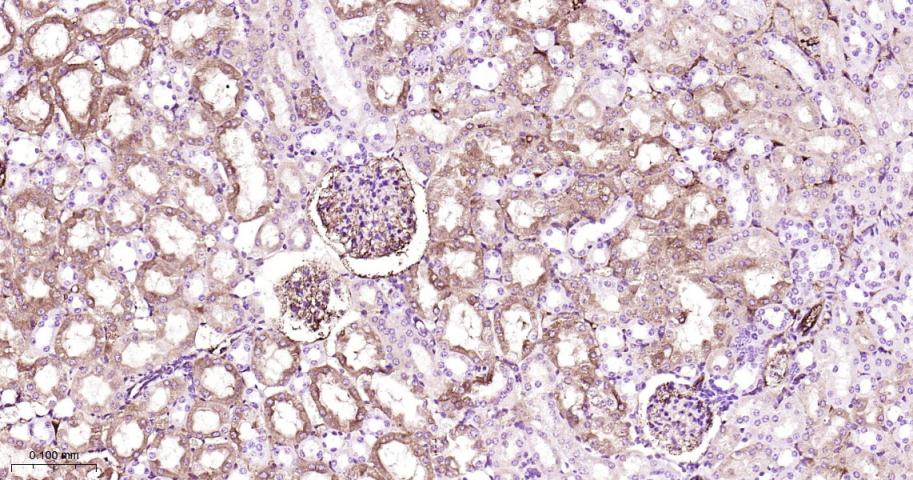
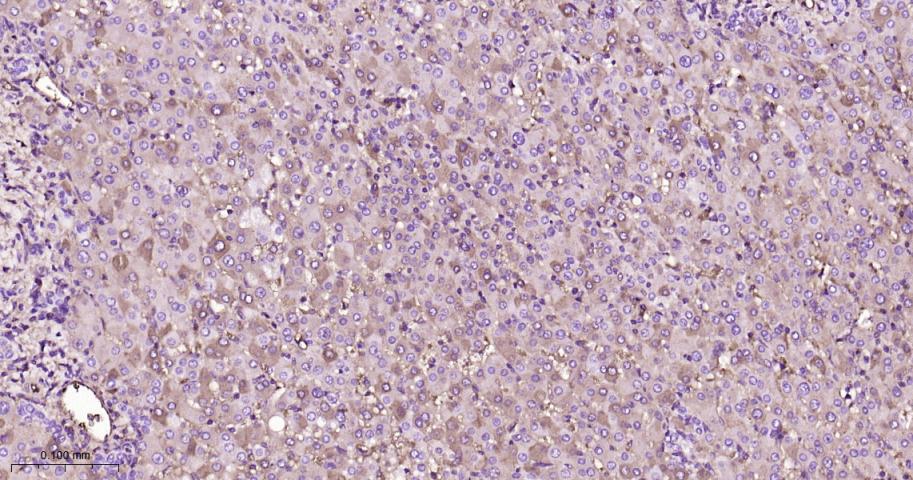
产品应用
| 应用 | 已检合格种属 | 预测种属 | 推荐稀释比例 |
|---|---|---|---|
| WB | Human | Mouse | 1:500-2000 |
| IHC-P | Human, Mouse | 1:50-200 | |
| IHC-F | Human, Mouse | 1:50-200 | |
| IF | Human, Mouse | 1:50-200 |
交叉反应
交叉反应: Human, Mouse
相关产品
暂无相关产品
同靶标产品
相关文献
提示: 发表研究结果有使用 bsm-61232R 时请让我们知道,以便我们可以引用参考文章。作为回馈,资料提供者将获得我们送上的小礼品。