Huntingtin Recombinant Rabbit mAb (一抗) - WB,IHC-P,IHC-F,IF,Flow-Cyt | Bioss
Rrmab?兔单抗
货号:bsm-54305R
产品详情
相关标记
相关产品
相关文献
常见问题
概述
产品编号
bsm-54305R
产品类型
重组兔单抗
英文名称
Huntingtin Recombinant Rabbit mAb
中文名称
神经性舞蹈病蛋白重组兔单抗
英文别名
HD; IT15; LOMARS; C430023I11Rik; Hdh; HD_HUMAN; HTT; Huntington disease protein (HD protein); HD_MOUSE; Huntington disease protein homolog (HD protein homolog); HD_RAT; huntingtin; huntingtin (Huntington disease)
抗体来源
Rabbit
免疫原
A synthesized peptide derived from human Huntingtin: 1-47
亚型
IgG
性状
Liquid
纯化方法
affinity purified by Protein A
克隆类型
Recombinant
克隆号
1F10
理论分子量
347 kDa
检测分子量
350 kDa
浓度
1mg/ml
储存液
0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol.
SWISS
Gene ID
保存条件
Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles.
注意事项
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
数据库链接
背景资料
Huntingtin is a protein that contains a polyglutamine region. When the number of glutamine repeats exceeds 35, the gene encodes a version of Huntingtin that leads to Huntington’s disease (HD). When the polyglutamine stretch is mutated, Huntingtin acts within the nucleus to induce neurodegeneration by a cell-specific apoptotic mechanism. Loss of Huntingtin activity is unlikely to be the cause of HD, and it has been proposed that the expanded glutamine repeat region may induce an abnormal interaction between the mutant protein and other cellular proteins. Huntingtin interacts with a variety of proteins including HAP1, glyceraldehyde phosphate dehydrogenase (GAPDH) and HIP1.
基因名
HTT
蛋白名
Huntingtin
亚基
Binds SH3GLB1 (By similarity). Interacts through its N-terminus with PRPF40A. Interacts with PQBP1, SETD2 and SYVN. Interacts with PFN1.
亚细胞定位
Cytoplasm. Nucleus. The mutant Huntingtin protein colocalizes with AKAP8L in the nuclear matrix of Huntington's disease neurons.
组织特异性
Expressed in the brain cortex (at protein level). Widely expressed with the highest level of expression in the brain (nerve fibers, varicosities, and nerve endings). In the brain, the regions where it can be mainly found are the cerebellar cortex, the neocortex, the striatum, and the hippocampal formation.
翻译后修饰
Cleaved by apopain downstream of the polyglutamine stretch. The resulting N-terminal fragment is cytotoxic and provokes apoptosis.
Forms with expanded polyglutamine expansion are specifically ubiquitinated by SYVN1, which promotes their proteasomal degradation.
Forms with expanded polyglutamine expansion are specifically ubiquitinated by SYVN1, which promotes their proteasomal degradation.
疾病
Defects in HTT are the cause of Huntington disease (HD) [MIM:143100]. HD is an autosomal dominant neurodegenerative disorder characterized by involuntary movements (chorea), general motor impairment, psychiatric disorders and dementia. Onset of the disease occurs usually in the third or fourth decade of life and symptoms progressively worsen leading to death in 10 to 20 years. Onset and clinical course depend on the degree of poly-Gln repeat expansion, longer expansions resulting in earlier onset and more severe clinical manifestations. HD affects 1 in 10,000 individuals of European origin. Neuropathology of Huntington disease displays a distinctive pattern with loss of neurons, especially in the caudate and putamen (striatum).
相似性
Belongs to the huntingtin family.
Contains 10 HEAT repeats.
Contains 10 HEAT repeats.
功能
May play a role in microtubule-mediated transport or vesicle function.
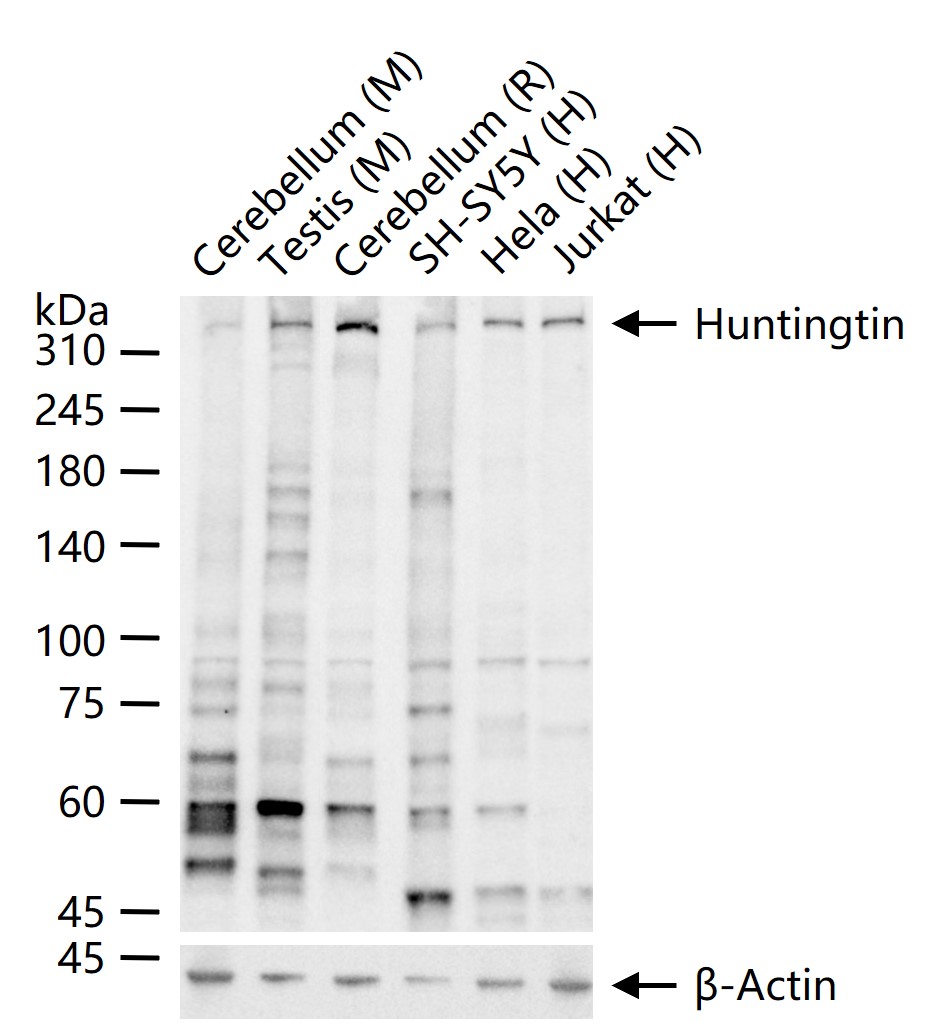
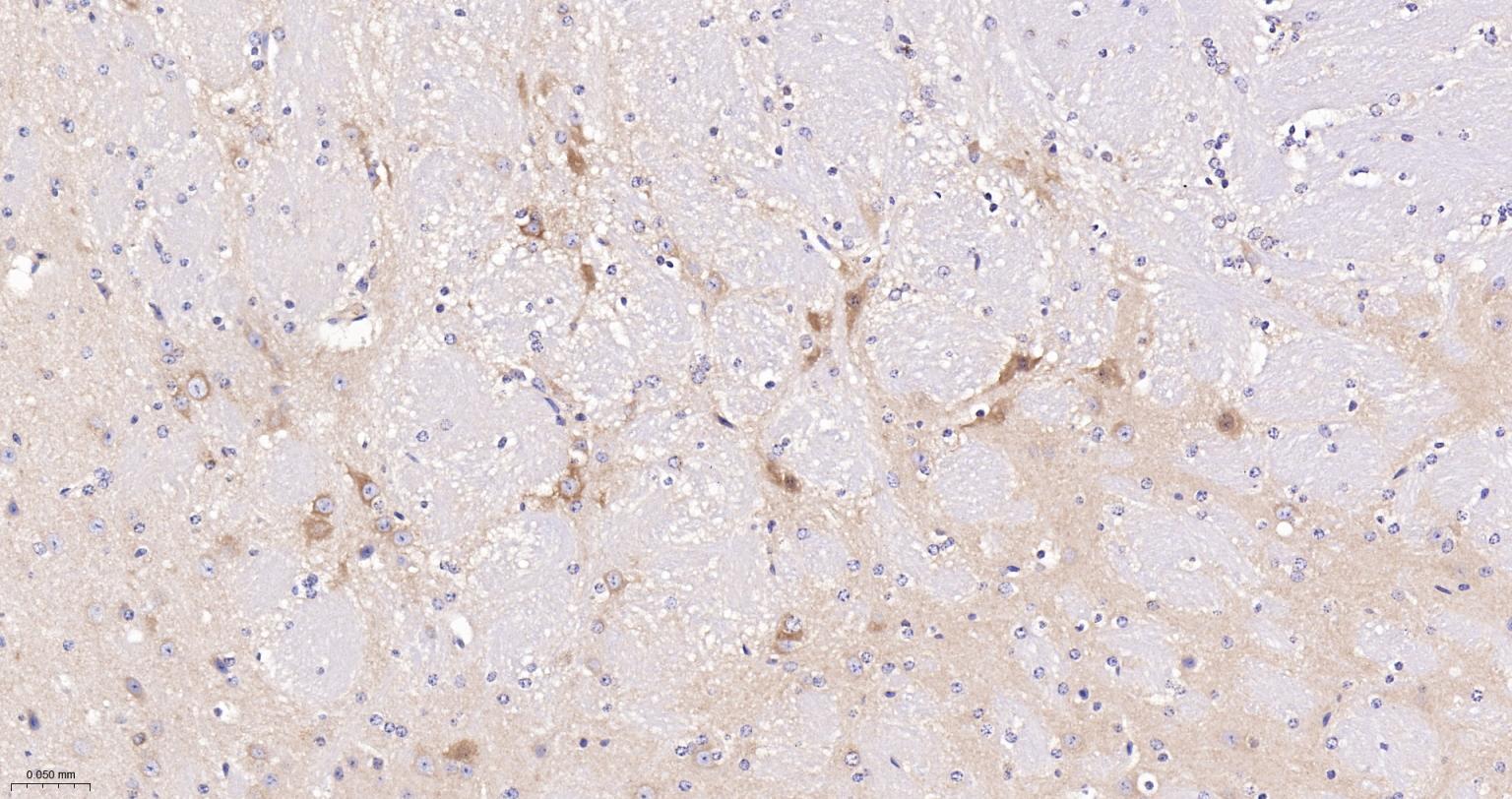
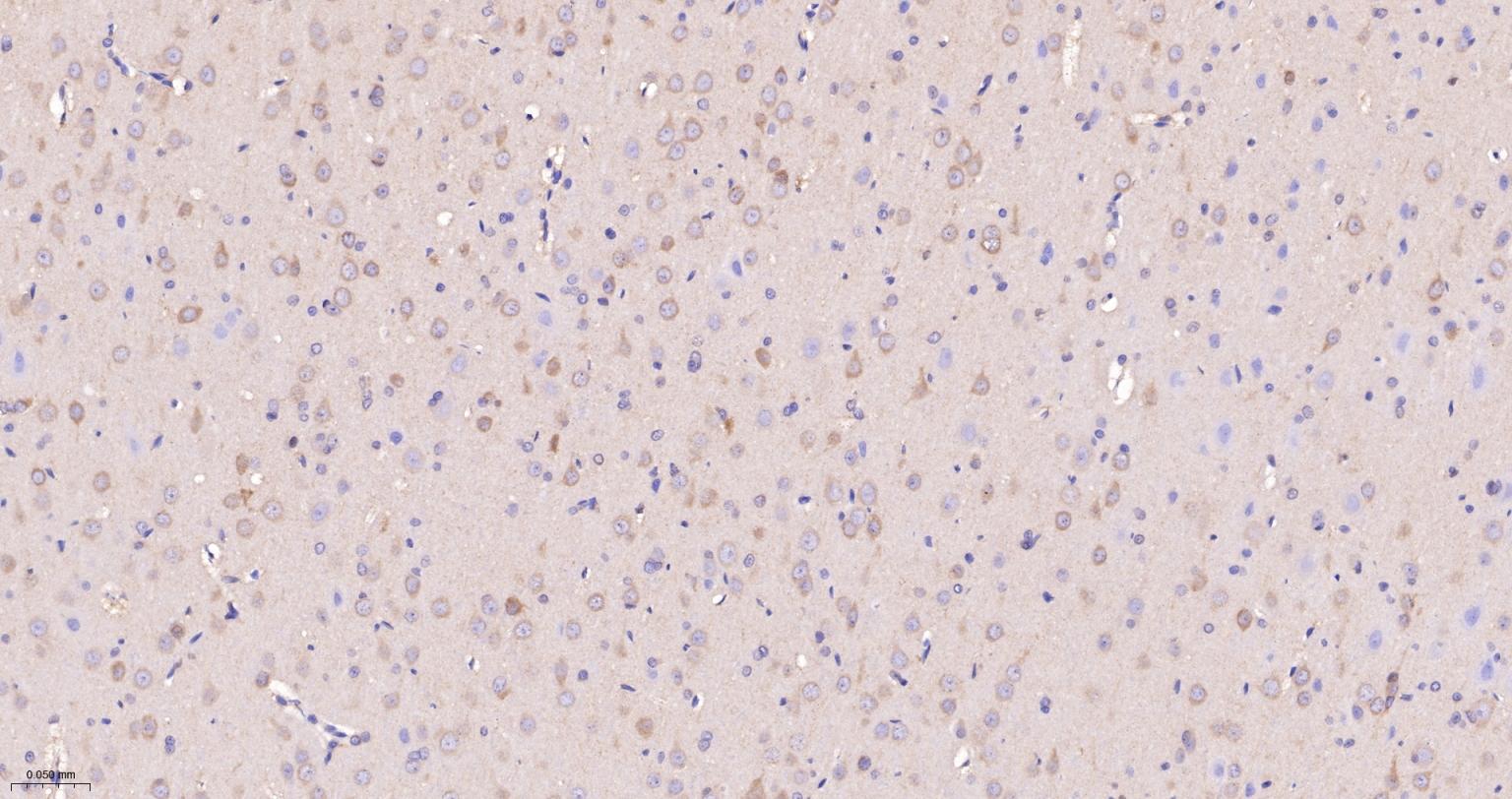
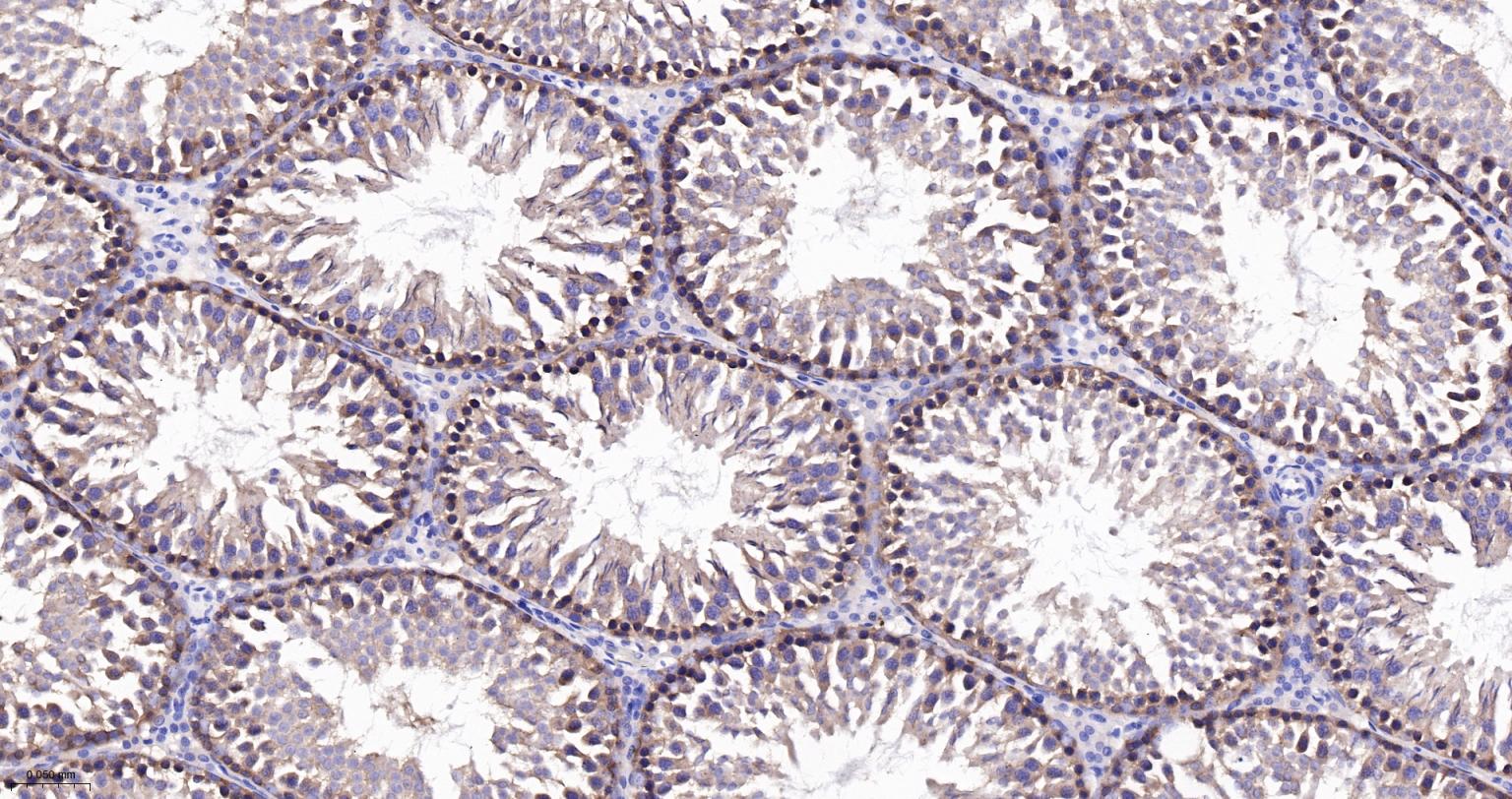
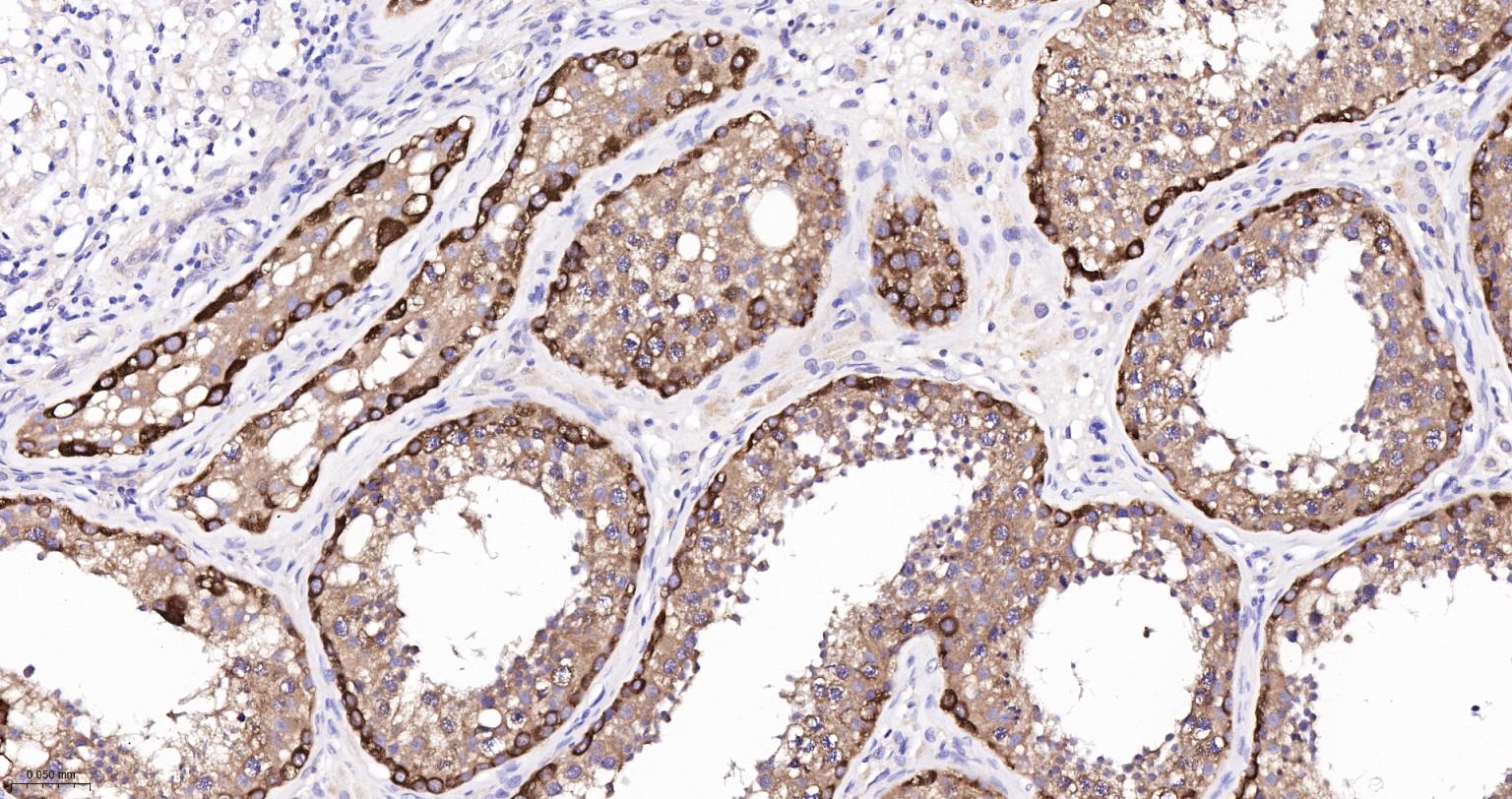
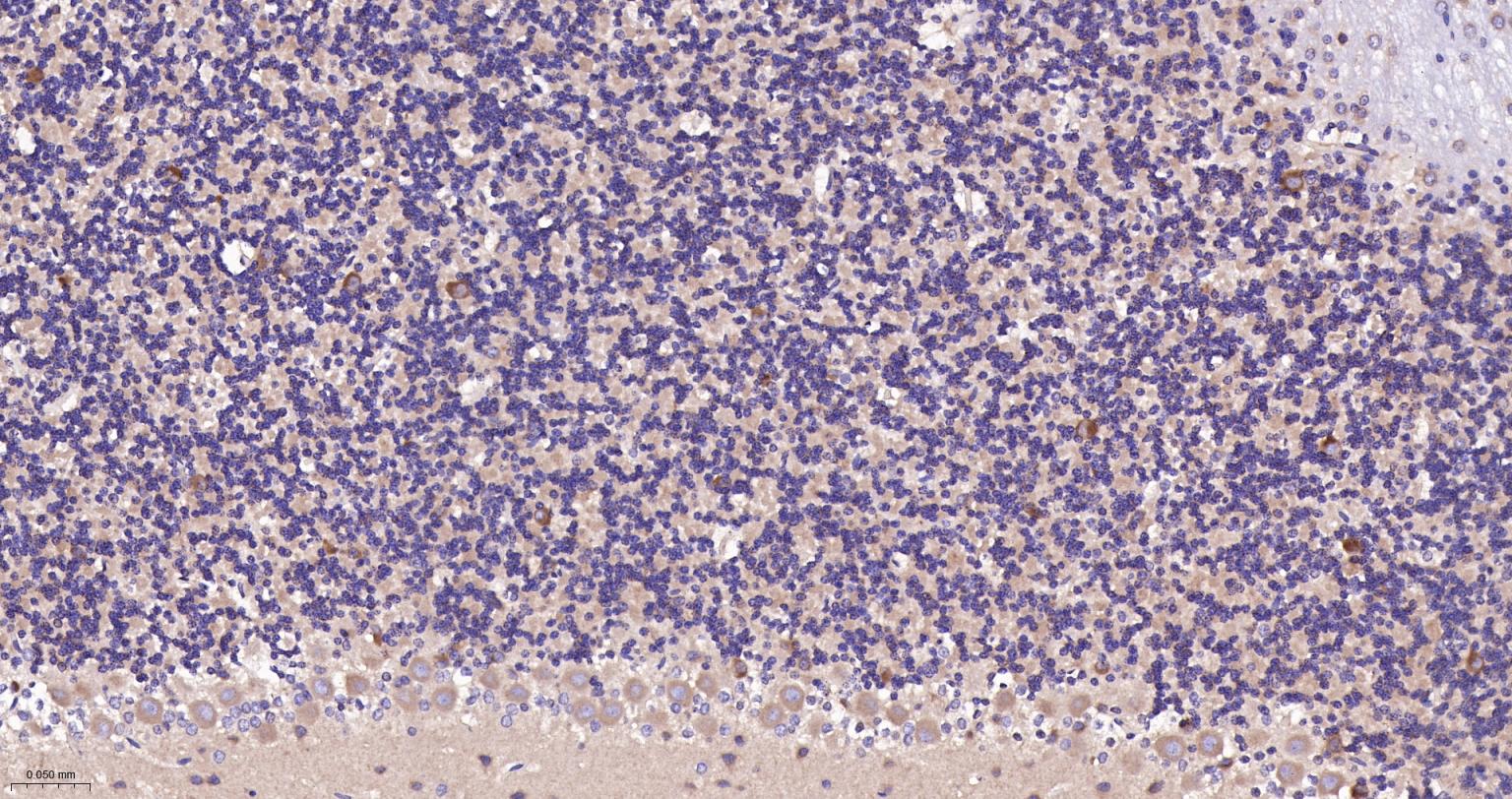
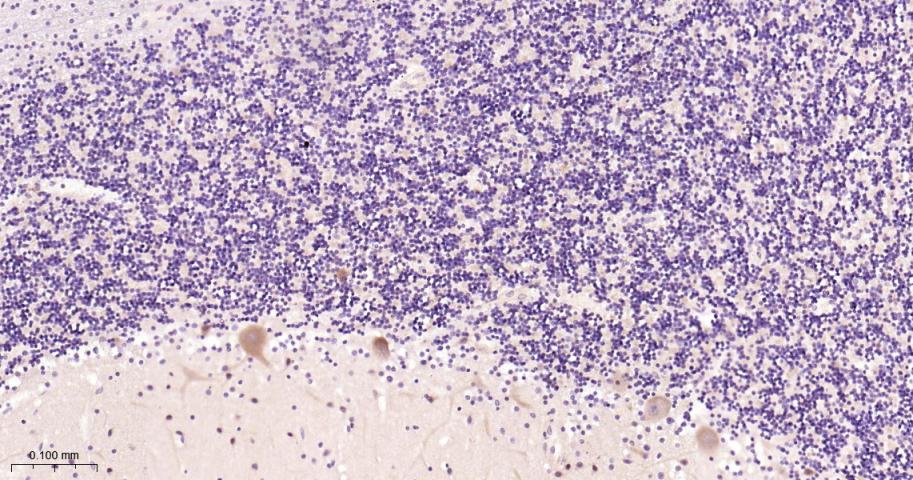
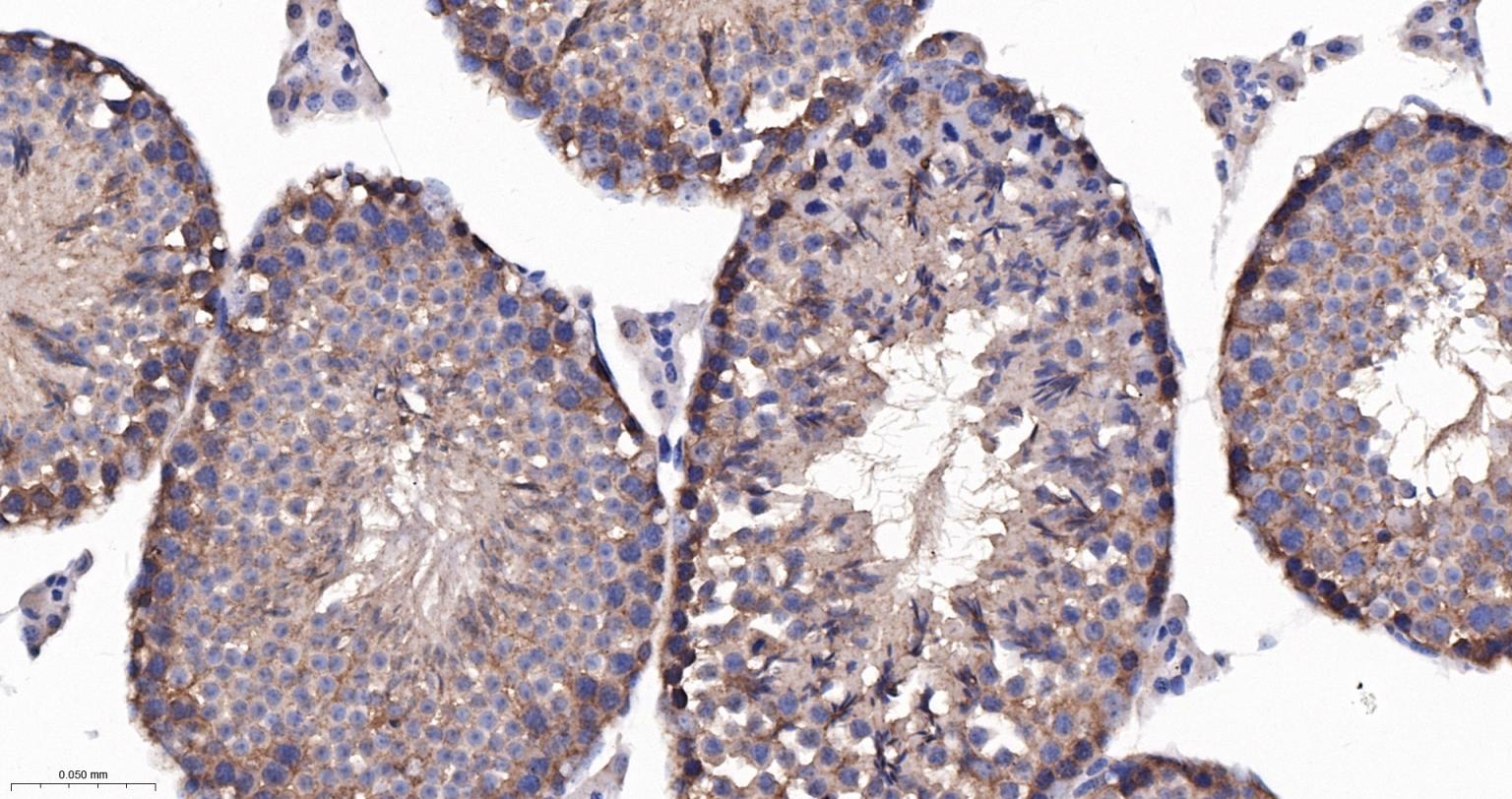
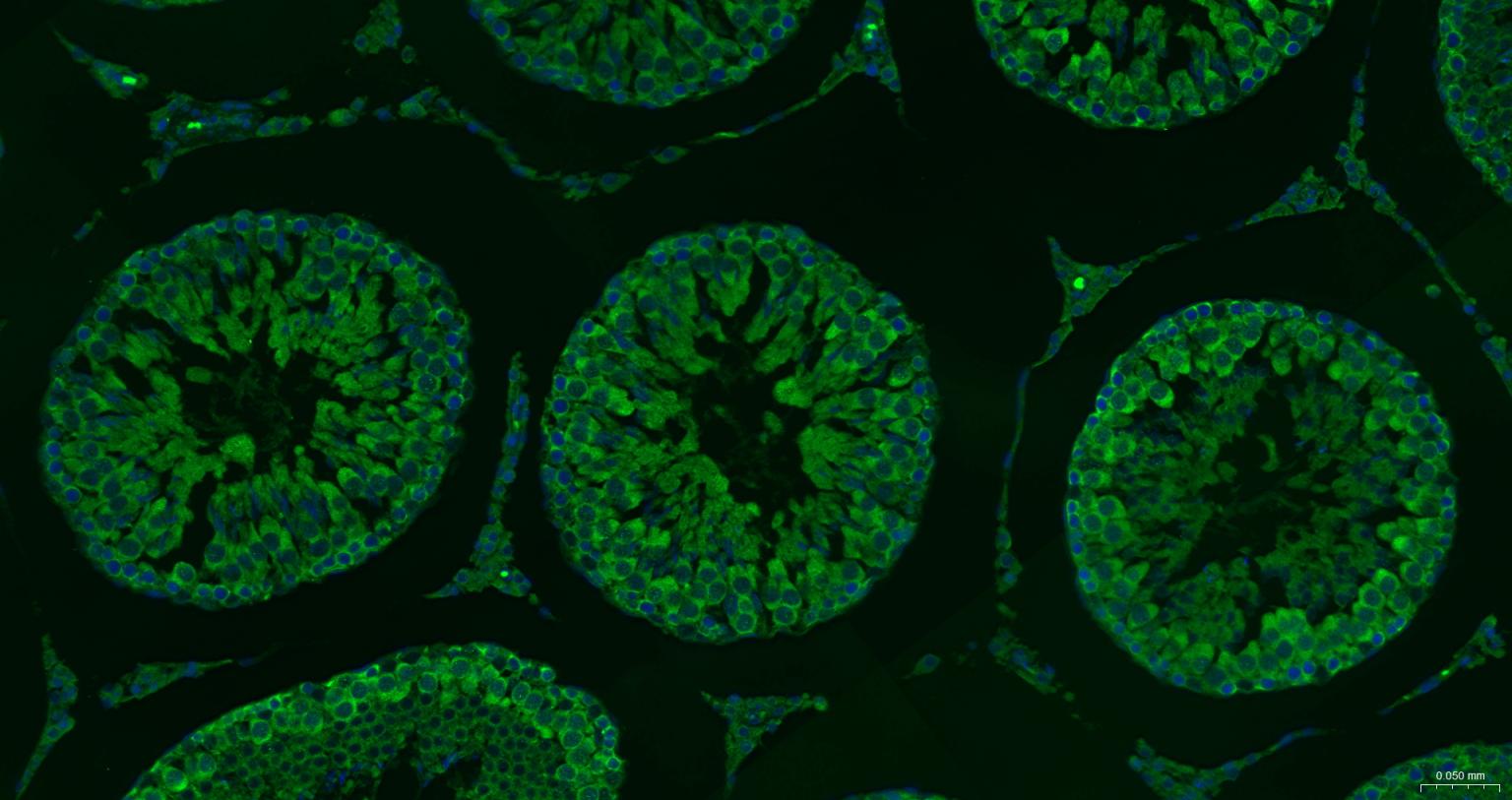
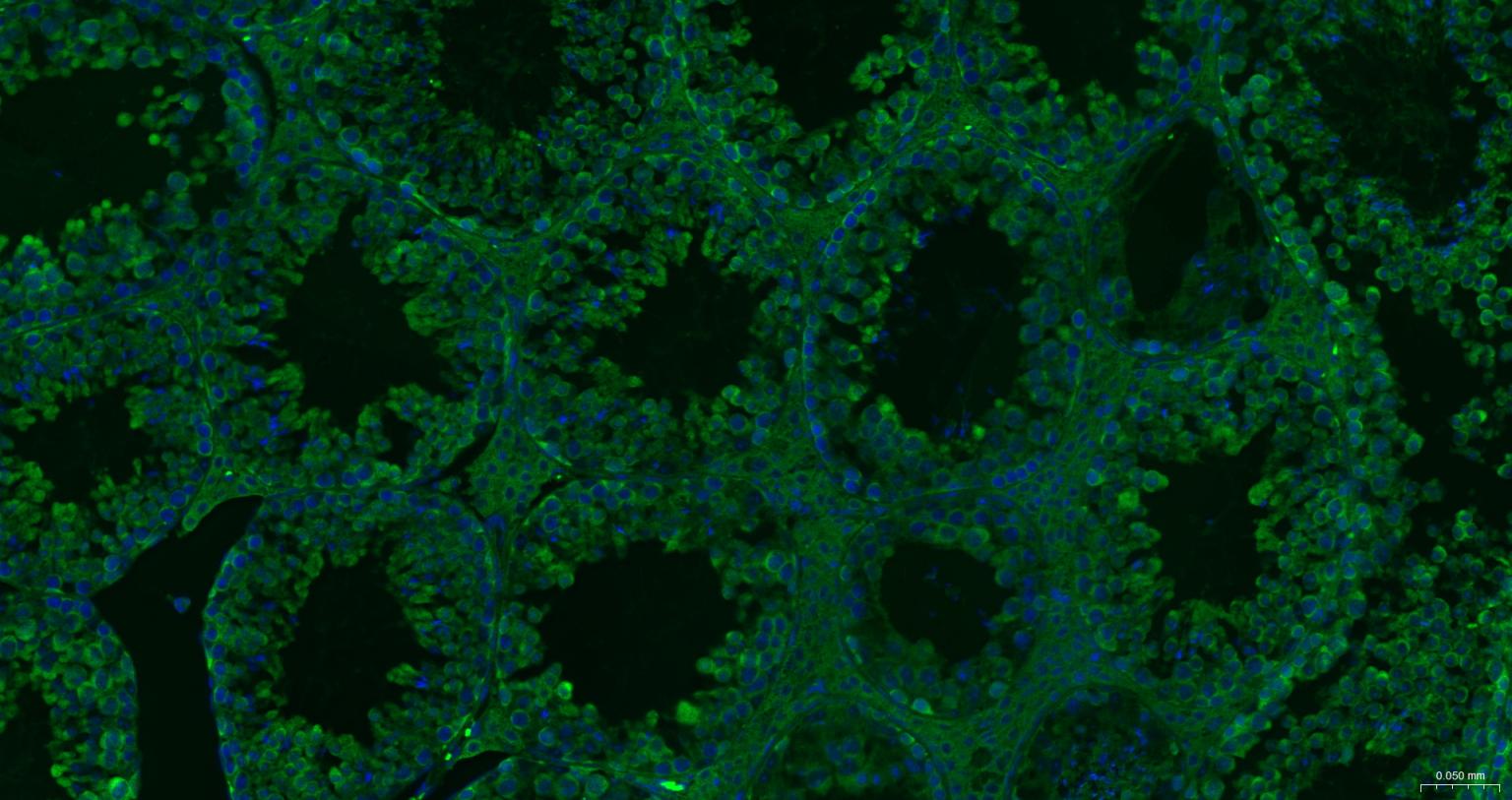
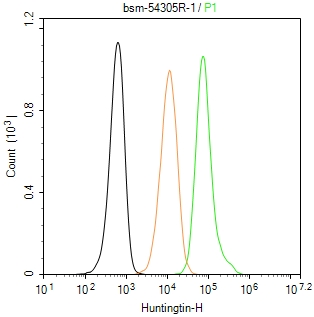
产品应用
| 应用 | 已检合格种属 | 预测种属 | 推荐稀释比例 |
|---|---|---|---|
| WB | Human, Mouse, Rat | 1:1000-2000 | |
| IHC-P | Human, Mouse, Rat | 1:100-500 | |
| IHC-F | Human, Mouse, Rat | 1:100-500 | |
| IF | Human, Mouse, Rat | 1:100-500 | |
| Flow-Cyt | Human | Mouse, Rat | 1μg/Test |
交叉反应
交叉反应: Human, Mouse, Rat
相关产品
暂无相关产品
同靶标产品
相关文献
提示: 发表研究结果有使用 bsm-54305R 时请让我们知道,以便我们可以引用参考文章。作为回馈,资料提供者将获得我们送上的小礼品。